FDA's 30 GLP-1 Warning Letters: The Largest Compounding Crackdown in Over a Decade
On March 3, 2026, the FDA issued 30 warning letters to telehealth companies selling compounded GLP-1s. Here's what happened, why it matters, and what QA teams should do now.

FDA's 30 GLP-1 Warning Letters: The Largest Compounding Crackdown in Over a Decade
On March 3, 2026, the FDA issued 30 warning letters to telehealth companies marketing compounded GLP-1 products. It was the single largest coordinated enforcement action against compounders since the agency overhauled compounding oversight after the 2012 New England Compounding Center (NECC) meningitis outbreak.
This was not a one-off. It was the second wave in six months. And it signals a fundamental shift in how the FDA polices the compounding sector.
What Happened on March 3
The FDA sent warning letters to 30 telehealth companies for making false or misleading claims about compounded semaglutide, tirzepatide, and in some cases liraglutide products on their websites. Companies named in reporting include Kin Meds, GoodGirlRx, WeightCare, and PharmaZee — largely smaller online wellness clinics operating direct-to-consumer models.
FDA Commissioner Marty Makary framed the action bluntly: "It's a new era. We are paying close attention to misleading claims being made by telehealth and pharma companies across all media platforms — and taking swift action."
The letters cited two primary categories of violations:
1. Implying sameness with FDA-approved products. Companies marketed compounded semaglutide and tirzepatide in ways that suggested equivalence with Ozempic, Wegovy, Mounjaro, and Zepbound. Compounded drugs are not FDA-approved. They are not generics. The agency has been clear: claiming or implying otherwise violates sections 502(a) and 502(bb) of the Federal Food, Drug, and Cosmetic Act (FDCA).
2. Obscuring product sourcing. Telehealth firms branded compounded products with their own names or trademarks without disclosing the actual compounding pharmacy, implying they manufactured the drugs themselves.
The FDA threatened "legal action without further notice" for companies that fail to correct.
The GenoGenix Case: When Marketing Problems Become Patient Safety Problems
One recipient stands out. GenoGenix LLC received a separate, more detailed warning letter (dated January 20, 2026) that went beyond marketing claims into manufacturing failures.
FDA inspectors found GenoGenix was repackaging semaglutide, tirzepatide, and retatrutide — the last being a next-generation GLP-1 still in clinical development at Eli Lilly and not yet approved by the FDA. The company was also compounding 5-amino-1-methylquinolinium iodide (5-Amino-1MQ) and nicotinamide adenine dinucleotide (NAD+), both ineligible for exemptions under section 503B of the FDCA.
The consequences were not theoretical. Three patients ended up in the emergency room after receiving a compounded NAD+ product from GenoGenix. Symptoms included low blood pressure, uncontrollable shaking, shivers, and body aches. An unopened vial from the same lot tested positive for excessive bacterial endotoxins.
Inspectors documented a litany of manufacturing failures:
- Products prepared, packed, or held in unsanitary conditions
- Personnel failing to disinfect materials before aseptic processing
- Staff exposing bare skin during sterile compounding
- Plant design that could compromise cleanroom integrity
- Missing written procedures for critical operations
GenoGenix is not just a marketing story. It is a patient safety case study — and a preview of where enforcement is heading.
This Is Not New: The NECC Shadow
The compounding industry has been here before. In September 2012, contaminated methylprednisolone acetate injections from the New England Compounding Center in Framingham, Massachusetts triggered a nationwide fungal meningitis outbreak. By the time the FDA's investigation concluded, 64 people were dead and 798 had been sickened across 20 states.
The NECC disaster exposed a regulatory gap. Traditional compounding pharmacies (operating under section 503A of the FDCA) were state-regulated and prepared drugs for individual patients with prescriptions. Large-scale compounders like NECC had grown into de facto manufacturers — shipping products interstate in bulk — while claiming the regulatory exemptions of a corner pharmacy.
Congress responded with the Drug Quality and Security Act (DQSA) in November 2013. The law created section 503B, establishing a new category of "outsourcing facilities" that could compound without individual prescriptions but had to register with the FDA, submit to inspections, report adverse events, and follow current good manufacturing practice (cGMP) requirements.
The framework was sound. Enforcement was slow. For over a decade, 503B oversight was largely reactive — the FDA investigated after problems surfaced. The GLP-1 compounding boom, fueled by shortages and consumer demand, exposed how much of the sector still operated in gray space.
The Six-Month Escalation
The March 3 letters did not come out of nowhere. The timeline tells the story:
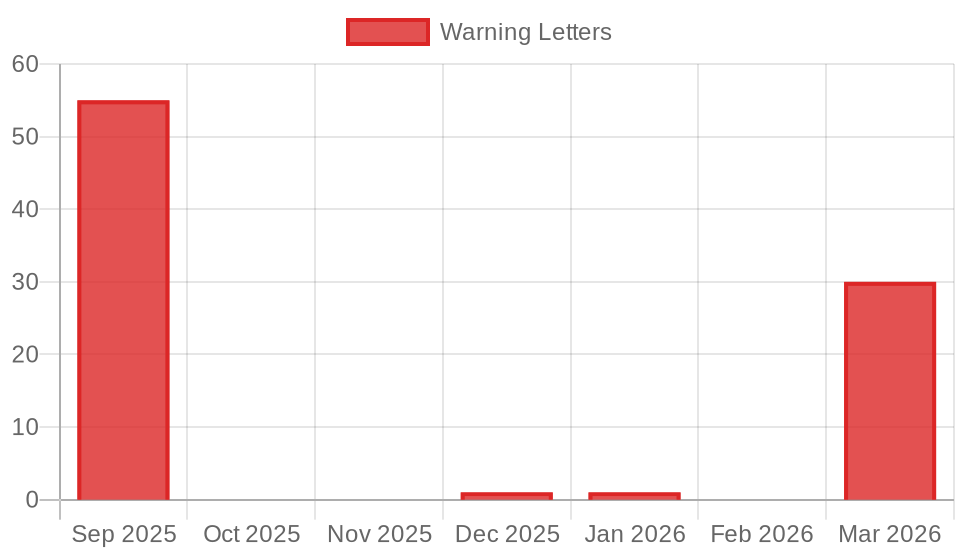
- October 2024: FDA removed tirzepatide (Mounjaro/Zepbound) from the drug shortage list after Eli Lilly stabilized supply.
- February 2025: FDA declared the semaglutide (Ozempic/Wegovy) shortage resolved, removing it from the shortage list.
- September 16, 2025: FDA published over 55 warning letters to GLP-1 compounders and online sellers — the first major wave. Hims & Hers Health received its letter on September 9, 2025.
- December 12, 2025: FDA issued a warning letter to MedisourceRx, the compounding pharmacy partnered with Hims & Hers.
- January 20, 2026: GenoGenix warning letter issued, citing manufacturing failures and adverse events.
- February 2026: Novo Nordisk and Eli Lilly began sending cease-and-desist letters to compounders directly.
- March 3, 2026: The 30 new warning letters dropped.
Over six months, the FDA sent more warning letters to compounding-related entities than it had in the preceding decade combined. Commissioner Makary said as much in the March 3 announcement.
The pattern is clear: reactive enforcement has shifted to proactive, systematic enforcement. The shortage rationale that shielded compounders is gone, and the agency is closing the gap.
The 503A vs. 503B Framework Under Pressure
Understanding the current crackdown requires understanding the two-track system Congress built after NECC.
Section 503A pharmacies compound drugs based on individual prescriptions. They are primarily state-regulated, exempt from FDA registration, and cannot advertise or promote specific compounded products. They fill a legitimate clinical need — patients with allergies, dosing requirements, or other needs that commercial products do not address.
Section 503B outsourcing facilities can compound without individual prescriptions and distribute to healthcare facilities. In exchange for this broader reach, they must register with the FDA, follow cGMP, submit to inspections, and report adverse events. They operate more like manufacturers but with compounding exemptions.
The GLP-1 boom blurred both categories. Telehealth companies partnered with 503A pharmacies to effectively mass-market compounded products — prescription-by-prescription, but at industrial scale and with consumer advertising that 503A was never designed to accommodate. Others used 503B facilities but marketed products in ways that implied FDA approval.
The March 3 letters targeted the telehealth front end — the marketing layer. But the enforcement net is widening to include the compounding pharmacies themselves, as the MedisourceRx and GenoGenix actions demonstrate.
What QA Teams at Compounding Facilities Should Do Now
If you operate or oversee quality at a compounding pharmacy — 503A or 503B — the signal from the FDA is unambiguous. Here is what needs attention immediately.
Review All Marketing Claims
Audit every customer-facing claim about your compounded products. Remove any language that implies equivalence to FDA-approved drugs. "Same active ingredient" is not the same as "same drug," and the FDA is drawing that line aggressively. Do not use brand names like Ozempic, Wegovy, Mounjaro, or Zepbound in promotional materials unless clearly disclaiming that your product is not FDA-approved and is not the branded product.
Validate Endotoxin Testing Protocols
The GenoGenix case is a direct warning. Bacterial endotoxin testing (BET) for injectable compounded products must be robust, documented, and traceable. If your facility compounds sterile injectables, confirm that every lot is tested per USP <85> (Bacterial Endotoxins Test) before release. Review your specifications — the limits, the methods, the equipment calibration records. An endotoxin failure in a released lot is a Class I recall waiting to happen.
Prepare for Inspection
The FDA's inspection cadence for 503B facilities is increasing. For 503A pharmacies, expect state boards to follow the FDA's lead. Ensure your facility can demonstrate:
- Current, complete batch records for every compounded product
- Environmental monitoring data for cleanrooms (viable and non-viable particulates)
- Personnel training records, including aseptic technique qualification
- Written procedures for every critical operation (the GenoGenix letter specifically flagged missing SOPs)
- Proper gowning, disinfection, and cleanroom behavior documentation
Reassess Your Product Portfolio
If you are compounding semaglutide or tirzepatide, the legal ground beneath you is eroding. Both drugs are off the shortage list. Novo Nordisk and Eli Lilly are sending cease-and-desist letters. The FDA's enforcement discretion periods have ended. Compounding these products outside of a legitimate 503A patient-specific prescription with a documented clinical need is increasingly indefensible.
If you are compounding substances ineligible for 503B exemptions — like the 5-Amino-1MQ and NAD+ cited in the GenoGenix letter — stop and consult regulatory counsel immediately.
The Broader Signal
The compounded GLP-1 market was estimated at over one million patients in 2025, according to Novo Nordisk. That market exists because branded drugs were unavailable or unaffordable. Both conditions are changing.
Novo Nordisk has stabilized semaglutide supply. Eli Lilly resolved the tirzepatide shortage in late 2024. Novo recently cut Wegovy's list price. Both companies are pursuing legal action against compounders. The FDA is providing the regulatory enforcement to match.
This is not just about GLP-1s. The FDA under Commissioner Makary has signaled a broader posture shift on compounding oversight — more proactive, more coordinated, and more willing to act at scale. The March 3 action hit 30 companies in a single day. The September 2025 wave hit more than 55. The pace is accelerating.
For compounding facilities operating within the rules — filling genuine clinical needs, maintaining quality systems, marketing honestly — nothing changes. The DQSA framework was built to protect that work.
For facilities that scaled up to meet a shortage and stayed after it ended, or that treated marketing regulations as optional, the window is closing. The FDA has made its intentions explicit. The enforcement actions are no longer warnings about what might happen. They are documentation of what is happening now.
The Takeaway
The FDA's March 3, 2026 enforcement action is the clearest signal yet that the compounding sector's post-shortage reckoning has arrived. Eighty-five warning letters in six months. Three patients in the ER from endotoxin contamination. A commissioner who publicly declared "a new era" of enforcement.
QA and regulatory teams at compounding facilities do not need to panic. They need to act. Audit marketing claims. Validate sterility testing. Document everything. And make an honest assessment of whether every product in your portfolio has a defensible regulatory basis.
The companies that do this work now will be the ones still operating a year from now.
Get this in your inbox every Friday.
Free FDA intelligence digest. No account required.